4-32
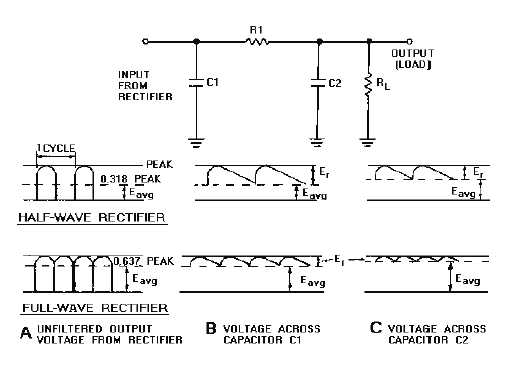
Figure 4-28.—RC filter and waveforms.
The RC filter in figure 4-28 consists of an input filter capacitor (C1), a series resistor (R1), and an
output filter capacitor (C2). (This filter is sometimes referred to as an RC pi-section filter because its
schematic symbol resembles the Greek letter
p
).
The single capacitor filter is suitable for many noncritical, low-current applications. However, when
the load resistance is very low or when the percent of ripple must be held to an absolute minimum, the
capacitor value required must be extremely large. While electrolytic capacitors are available in sizes up to
10,000 microfarads or greater, the large sizes are quite expensive. A more practical approach is to use a
more sophisticated filter that can do the same job but that has lower capacitor values, such as the RC
filter.
Views A, B, and C of figure 4-28 show the output waveforms of a half-wave and a full-wave
rectifier. Each waveform is shown with an RC filter connected across the output. The following
explanation of how a filter works will show you that an RC filter of this type does a much better job than
the single capacitor filter.
C1 performs exactly the same function as it did in the single capacitor filter. It is used to reduce the
percentage of ripple to a relatively low value. Thus, the voltage across C1 might consist of an average dc
value of +100 volts with a ripple voltage of 10 volts peak-to-peak. This voltage is passed on to the R1-C2
network, which reduces the ripple even further.
C2 offers an infinite impedance (resistance) to the dc component of the output voltage. Thus, the dc
voltage is passed to the load, but reduced in value by the amount of the voltage drop across R1. However,
R1 is generally small compared to the load resistance. Therefore, the drop in the dc voltage by R1 is not a
drawback.
Component values are designed so that the resistance of R1 is much greater than the reactance (XC)
of C2 at the ripple frequency. C2 offers a very low impedance to the ac ripple frequency. Thus, the ac